戳上方藍色字體「艾德研究部」了解最新資訊
核心事件:2026年4月30日,FDA腫瘤藥物諮詢委員會(ODAC)以6票反對、3票贊成的結果,不建議批准阿斯利康新一代口服SERD藥物camizestrant聯合CDK4/6抑制劑用於HR+/HER2-伴ESR1突變晚期乳腺癌的一線治療。否決核心在於試驗設計缺陷(基於ctDNA分子進展提前換藥,無法直接證明優於影像學進展後換藥)、缺乏長期OS數據、對照組不允許交叉用藥導致偏倚,以及尚無同類監管先例。
對阿斯利康的直接影響:雖然該適應症對公司整體營收敞口有限,但camizestrant曾被寄予年銷售峰值超50億美元的預期,此次否決使美國市場准入受阻。不過,歐盟EMA CHMP已於2026年5月採納積極意見,預計下半年在歐盟獲批。SERD賽道競爭格局分化:禮來憑藉先發優勢暫時領跑,羅氏轉向早期輔助治療。
行業監管標準全面抬升:FDA釋放明確轉向信號——不再接受僅憑替代終點(如PFS)或分子標誌物驅動的“早期干預”策略作為獲批依據,必須提供直接臨床獲益證據(如OS改善)。創新性試驗設計(如序貫治療對比)在對照組設置、交叉用藥規則、偏倚防控方面面臨從嚴審查,試驗架構、分組邏輯、交叉用藥、偏倚防控四大維度標準全面收緊。
行業面臨長期正向變革:倒逼行業回歸臨床本質,推動臨床試驗設計前置優化、優先規劃OS硬終點、加強早期監管溝通、提升真實世界證據(RWE)的應用價值,利於行業健康可持續發展。
2026 年 4 月 30 日,美國 FDA 腫瘤藥物諮詢委員會 (ODAC) 以6 票反對、3 票贊成的結果,不建議批准阿斯利康 (AstraZeneca) 在研新藥 camizestrant 聯合 CDK4/6 抑制劑用於特定類型乳腺癌的治療。這一非約束性投票結果對 FDA 最終審批具有重要參考意義,使該藥物獲批前景變得黯淡。
藥物與適應症背景
Camizestrant屬於新一代口服選擇性雌激素受體降解劑(SERD) 和完全雌激素受體拮抗劑,阿斯利康研發的潛在重磅乳腺癌藥物。該藥物的適應症為聯合呱柏西利、瑞波西利或阿貝西利等CDK4/6 抑制劑,用於 HR 陽性、HER2 陰性晚期乳腺癌患者在一線治療中檢測到 ESR1 突變時的治療,且無需等到影像學進展;臨床依據為基於III 期 SERENA-6 試驗 (NCT04964936) 數據,該試驗顯示與繼續使用芳香化酶抑制劑 (AI)+CDK4/6 抑制劑相比,提前轉換為 camizestrant+CDK4/6 抑制劑可顯著延長 PFS (無進展生存期) 和 PFS2 (至第二次進展或死亡時間)。
否決核心原因
美國FDA 腫瘤藥物諮詢委員會 (ODAC)的主要擔憂集中在以下幾點:
後續進展
阿斯利康表示將繼續與FDA 合作,完成 NDA 審查,並計劃將在5月29日至6月2日舉行的2026年美國臨床腫瘤學會 (ASCO) 年會上公佈最終PFS2 結果。該公司同時強調,camizestrant 在其他適應症 (如 AI 治療失敗後的二線治療) 的開發不受影響。
2.1. 對阿斯利康的直接影響
1)股價波動:圍繞在研新藥Camizestrant的在研進展,阿斯利康股價經歷了三輪顯著的波動,分別對應:2025 年 2 月(中期分析積極結果)、2025 年 10 月(賣方分析師降級) 以及 2026 年 4 月—5 月(FDA 諮詢委員會投票結果出爐),反映出市場對該藥物商業化前景的擔憂。
第一輪,2025年2月,中期分析“實質性積極意外”推動股價上漲。2025 年 2 月 26 日,阿斯利康宣佈 SERENA-6 III 期試驗的中期分析達到 PFS 主要終點,即對於合併 ESR1 突變的晚期 HR 陽性乳腺癌患者,Camizestrant 一線治療可使患者無進展生存期(PFS)得到具有統計學差異與臨床價值的明顯改善,研究將繼續按計劃進行。當天,阿斯利康股價收報203.98,小幅下跌 0.88%。這或源於生物制藥行業“利好兌現(Sell the News)”的常見市場反應,且該消息基本符合業界預期。
儘管當日微跌,但該消息被業界定性為"實質性積極意外"(material positive surprise)。阿斯利康曾預測,camizestrant有望於今年內獲得美國監管機構批准,其年銷售額峰值預計將突破50億美元。這一臨床進展奠定了該候選藥物在阿斯利康乳腺腫瘤管線中的核心地位,符合業界預期。
第二輪,2025 年 10 月——德銀罕見"賣出"評級引發市場疑慮。2025 年 10 月 16 日,德意志銀行分析師將阿斯利康評級從"持有"罕見下調至"賣出",對阿斯利康的藥物研發管線(尤其是乳腺癌療法)持更為審慎的看法。當日阿斯利康股價收報165.916,下跌1.18%。
第三輪,2026 年 4 月—5 月——FDA 諮詢委員會公佈負面投票結果。2026 年 4 月 30 日,FDA 腫瘤藥物諮詢委員會(ODAC)以 6 票反對、3 票贊成的投票結果,否決了 camizestrant 聯合 CDK4/6 抑制劑用於一線治療 HR+/HER2- 伴 ESR1 突變晚期乳腺癌的獲益風險譜。當日,阿斯利康股價小幅收漲1.17%。這或主要源於:一方面,市場對結果已有預期:Jefferies 分析師 Michael Leuchten 指出,在 FDA 公佈的備審簡報檔(briefing documents)發佈後,這一負面投票"並不令人意外",因此市場提前消化了部分負面資訊;另一方面,此適應症對整體業績敞口有限:該特定適應症(伴ESR1 突變的晚期乳腺癌一線治療)僅占阿斯利康整體產品管線和 2030 年約 800 億美元銷售目標的很小一部分。Jefferies 認為,這一消息造成的任何短期弱勢都應被視為買入機會。花旗亦在研報中指出,委員的反對意見並非基於藥物毒性擔憂,這意味著該藥在更廣泛的後續試驗中仍具備競爭力。
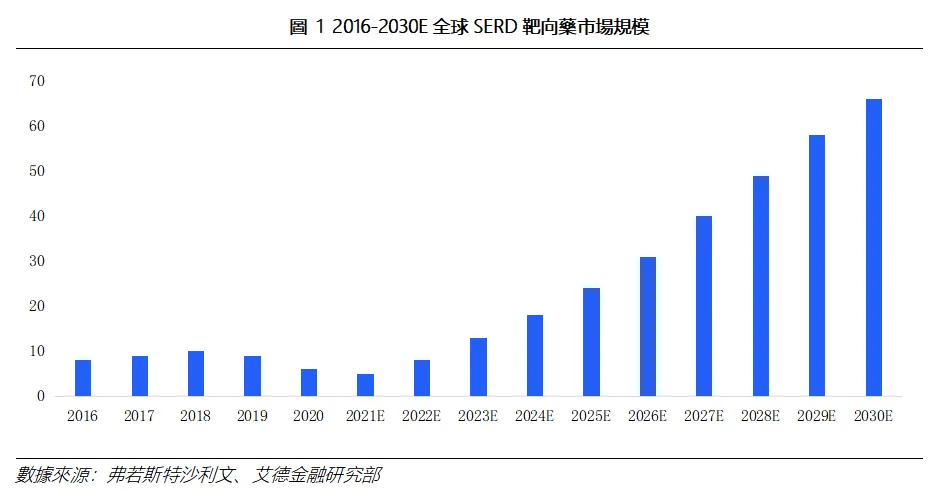
2)研發管線調整:雌激素受體調節劑(SERD)賽道的競爭格局或因阿斯利康此次的監管受挫悄然分化。SERD領域早已不再是單一目標的競賽。全球SERD市場正在高速增長。據弗若斯特沙利文預測,該市場將從2025年的約24.0億美元增長至2030年的約66.0億美元,年複合增長率高達22.4%。
雖然阿斯利康在核心管線進度上遭遇監管打擊,但憑藉其全球化的佈局和在研管線的深度,其並未完全出局。2026年5月22日,EMA人用藥品委員會(CHMP)正式採納了camizestrant(歐盟商品名 Etcamah)聯合CDK4/6抑制劑的積極審評意見。該療法用於治療成人雌激素受體(ER)陽性、人表皮生長因數受體2(HER2)陰性局部晚期或轉移性乳腺癌,適用條件為:患者在一線內分泌治療聯合CDK4/6抑制劑期間檢測出ESR1突變,且尚未發生影像學疾病進展。可聯合的CDK4/6抑制劑包括呱柏西利(palbociclib)、瑞博西尼(ribociclib)和阿貝西利。本次CHMP積極意見適用集中審評程式(Centralised Procedure),批准後可在歐洲經濟區(EEA,含歐盟27國及挪威、冰島、列支敦士登)全面上市。CHMP的積極意見將提交歐盟委員會(EC)作最終批准決定,通常這一流程耗時約2-3個月,預計2026年下半年可獲得正式上市許可。此次於FDA監管受挫也僅是在進軍美國市場的關鍵路徑上受到阻礙。
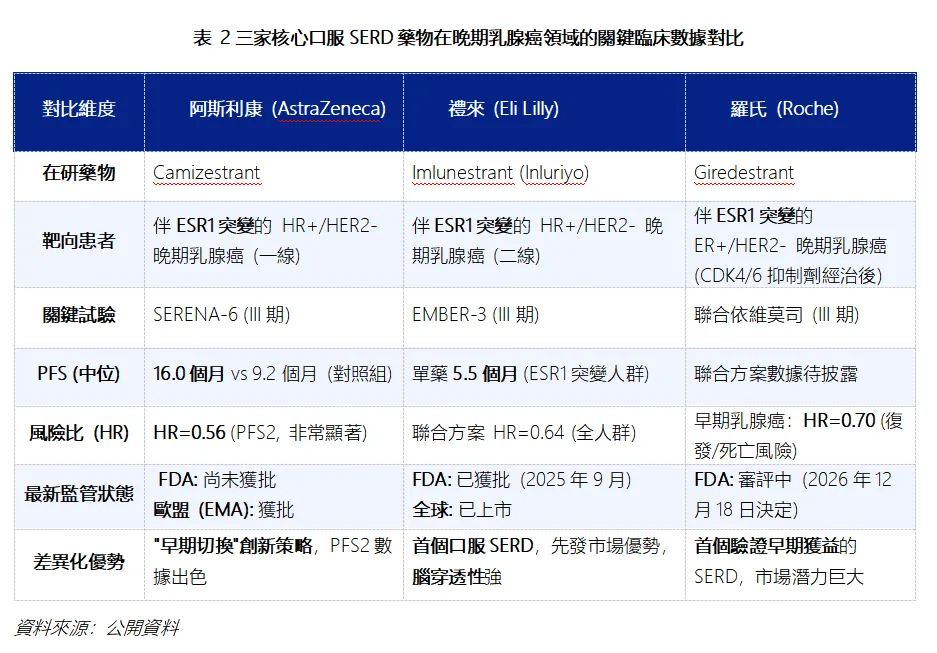
在美國市場內,已有禮來憑藉快速的市場准入暫時領跑,而羅氏則在早期治療階段展現出了差異化優勢。
從上表可以看出,禮來是先發者,其Imlunestrant 不僅獲批上市,還具有“強腦穿透性”的差異化特性,可能對存在腦轉移風險的患者更具吸引力;羅氏是差異化佈局者,雖然其 Giredestrant 在晚期乳腺癌一線治療中曾遭遇挫折,但最新的 lidERA III期研究顯示,在早期乳腺癌輔助治療中,能降低30%的復發或死亡風險,成為首個在早期階段驗證獲益的口服SERD,開闢了全新的廣闊市場;阿斯利康則憑藉 Camizestrant 在機制和策略上尋求突破。其 SERENA-6 試驗採用創新的“ctDNA指導下的早期切換”策略,在疾病影像學進展前就切換治療方案,在PFS及PFS2(HR=0.56)等終點上展現出顯著優勢。
而阿斯利康Camizestrant此次監管遭遇否決,顯然在阿斯利康進軍美國市場的關鍵路徑上設置了障礙,短期抬升了阿斯利康監管上的不確定性,驗證了其針對ESR1突變患者的“早期切換”創新策略的臨床價值並未獲得FDA諮詢委員會的完全認可。對於禮來,由於其Imlunestrant (Inluriyo) 於2025年9月獲批用於二線治療,目前是市場中唯一可及的口服SERD。阿斯利康的受挫,為禮來提供了寶貴的時間窗口,使其能夠從容地進行市場滲透和教育,而無直接的同代產品競爭壓力。對於羅氏,雖然曾於2025年早先時候遭遇過晚期適應症的失敗,其聚焦點已轉向更廣闊的早期輔助治療。2026年,其 lidERA III期試驗結果為陽性,驗證了口服SERD在早期乳腺癌中的巨大潛力。預計在2026年底前獲得FDA審批決定,若獲批,將為其在早期市場中建立先發優勢。
阿斯利康此次FDA監管層面的受挫,某種意義上來說,或許意味著在SERD口服藥甚至是在未來乳腺癌創新藥行業的競爭將從單純的臨床有效性,轉向商業策略、市場准入速度、全球市場佈局(尤其是歐盟和中國)以及在研管線的深度和廣度。
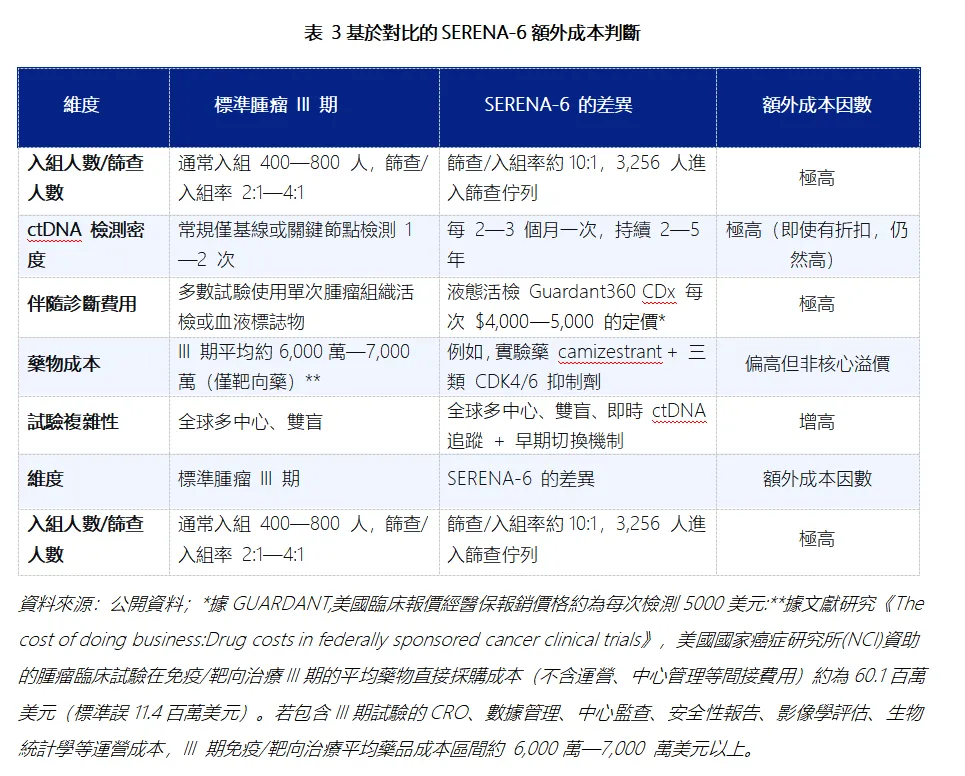
3)財務影響:首先,SERENA-6 試驗投入體量確實可觀,阿斯利康顯然對該藥寄予厚望。從臨床試驗的經濟學角度,SERENA-6作為一項全球多中心、包含常規週期液體活檢(ctDNA持續監測)機制的大型III期試驗,其運營難度和伴隨檢測成本均顯著高於典型的腫瘤III期試驗,啟動該項試驗的固定沉沒成本確實不容忽視。這主要體現在兩個方面,一個是超常規的大規模名義篩查(據SERENA-6試驗的公開分組數據,3,256人vs最終僅入組315人,有效篩查/入組比高達10:1);另一個是試驗期間要求的每 2—3 個月一次連續 ctDNA 監測,疊加全球多中心執行複雜度和高單價伴隨檢測成本。
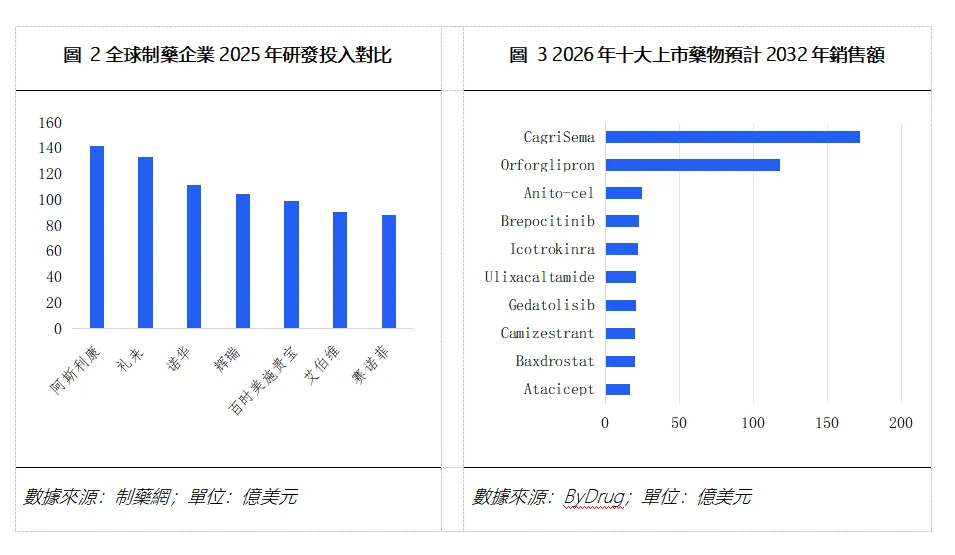
在資金投入層面,阿斯利康自身2025年全年研發投入高達142.32億美元,同比增長4%,占其營收比重約24.2%,在全球制藥企業中處於較高水準。SERENA-6作為其乳腺癌核心管線,在其中佔據顯著資源權重。
在財務預期層面,阿斯利康公司自身預測camizestrant的年銷售額峰值有望超過50億美元,被視為達成"2030年800億美元總營收目標"的關鍵動能之一。Evaluate Pharma則給出了更為保守的預測——2032年全球銷售額約20億美元,並將其列入"2026年最受期待的十大上市新藥"之一。
在更大戰略層面,阿斯利康設定了2030年實現800億美元營收的宏偉目標,且該藥物被納入其中。因此,"若最終未能獲批"確實可能對其整體業績節奏造成一定擾動。
2.2. 對行業層面的直接影響
由點及面,本次阿斯利康Camizestrant 被 FDA 腫瘤藥物諮詢委員會否決,並非單一藥企的專案失利,而是美國 FDA 向全球腫瘤創新藥行業釋放明確的監管風向轉變信號。否決的核心爭議(試驗設計缺陷、過度依賴替代終點、創新治療範式缺乏臨床獲益佐證、無監管先例),層層傳導至臨床試驗設計、審批標準、前沿技術落地、企業研發策略等全鏈條,短期推高行業風險,長期倒逼行業走向更嚴謹、更貼合臨床價值的研發邏輯。
2.2.1. 臨床終點與臨床試驗設計的監管標準全面抬升
分子標誌物指導的早期干預策略和“治療時機對比”類臨床試驗設計是當前腫瘤新藥研發的兩大主流方向。阿斯利康Camizestrant被FDA否決的事件或反映出臨床終點與臨床試驗設計的監管標準正全面抬升。
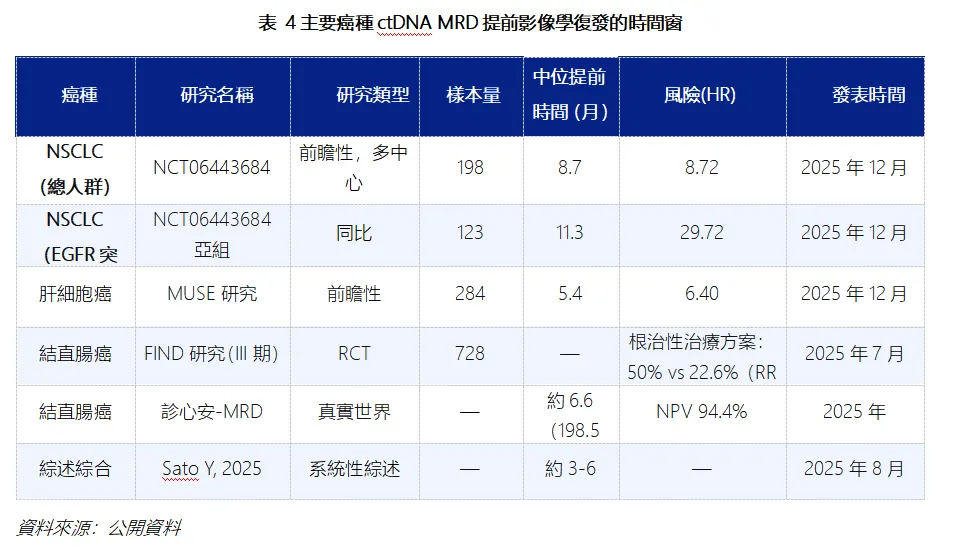
分子標誌物驅動的早期干預策略全面承壓。Camizestrant 申請的適應症,是依託ctDNA 檢測 ESR1 基因突變(分子進展),不等影像學發現病灶進展就提前更換治療方案,這是近幾年全球乳腺癌、實體瘤領域非常熱門的精准醫療探索方向。
FDA 及 ODAC 明確表態,不認可僅依靠分子標誌物陽性就啟動提前換藥的治療模式。過往行業普遍認為,ctDNA 等液體活檢能更早捕捉疾病變化、提升治療效果,因此大量藥企跟風佈局 “分子進展即干預” 的研發路線。
阿斯利康Camizestrant 的6比3的投票否決結果表明,在缺乏成熟OS數據且非傳統影像學進展後換藥的條件下,ESR1突變陽性檢測本身不足以成為批准依據。這意味著技術上可實現不等於臨床上有明確獲益,單純依靠基因或分子指標指導治療時序調整,無法成為獲批依據。
未來同類創新療法,不能只證明“分子指標改善”,必須拿出直接證據,證明該干預方式能給患者帶來實實在在的臨床收益。同時,由於該治療模式暫無監管先例,FDA 對這類 “範式創新” 會保持高度保守,進一步抬高准入門檻。據Fierce Biotech報導,FDA在會議期間即提出該試驗設計無法回答早期切換策略的長期獲益問題。
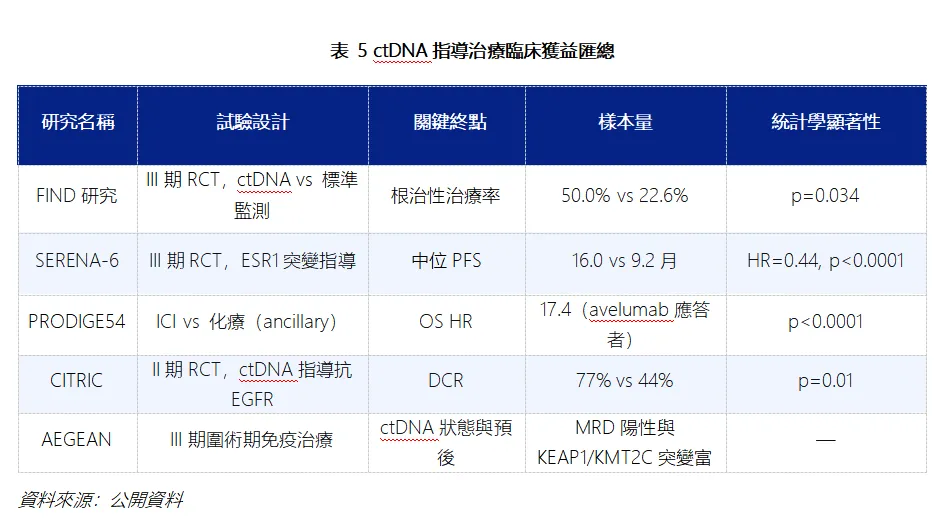
非傳統試驗設計、對照組設置成為審評核心紅線。阿斯利康III 期 SERENA-6 試驗屬於 “早用藥 vs 晚用藥”的序貫治療對比試驗,區別於傳統 “藥物 vs 標準療法 / 安慰劑” 的優效性試驗,本身結果解讀難度更大;疊加對照組患者疾病進展後不允許交叉使用 Camizestrant,被專家認定存在試驗偏倚。中國藥行業分析已明確指出,SERENA-6研究本身並非為回答PFS2與OS問題而設計,對照組患者在影像學進展後不允許交叉換組,這使兩組的比較失去了對等前提。
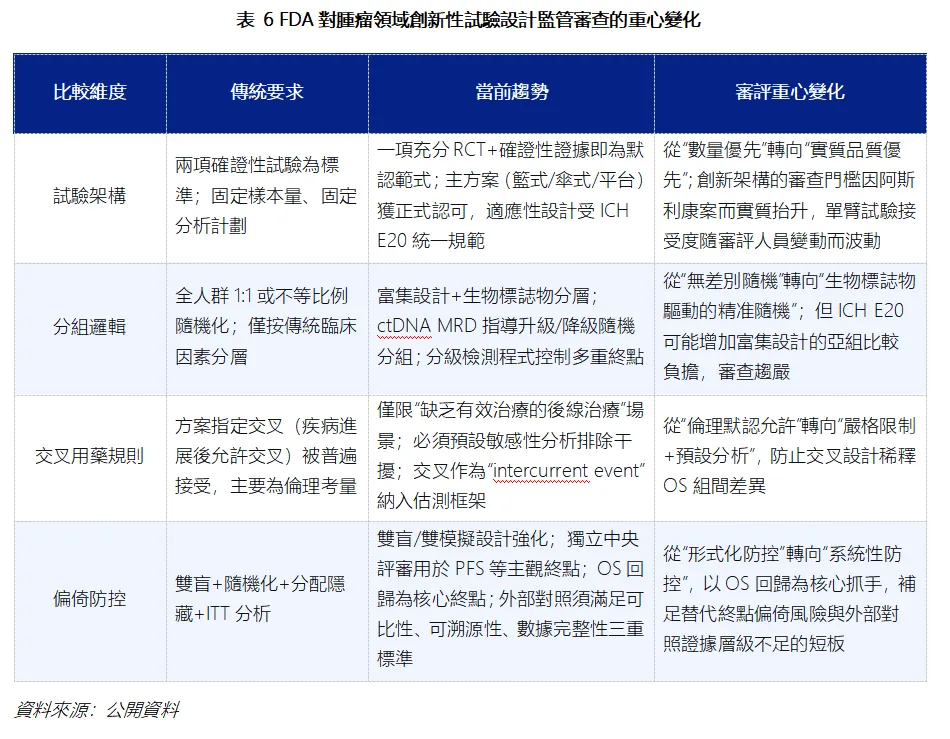
此前FDA 對腫瘤領域創新性試驗設計包容度較高,如今會對試驗架構、分組邏輯、交叉用藥規則、偏倚防控進行從嚴審查。這意味著FDA監管審查的重心出現了變化。試驗架構從“固定設計”走向“主方案+適應性設計”,但創新必須伴隨足夠的證據實質支撐;分組邏輯從“全人群隨機”走向“富集+分層”,但富集策略正面臨更嚴格的審查要求;交叉用藥從“倫理默認允許”走向“嚴格限制+預設分析”,以防止生存獲益被稀釋;偏倚防控則從“形式合規”走向“OS回歸+系統化防控”。
這或許意味著後續藥企開展“治療時機選擇”“序貫療法對比” 這類試驗時,儘量在方案設計階段就充分論證對照組合理性、規避結果偏倚,試驗設計的容錯空間大幅縮小。
2.2.2. 全球腫瘤創新藥審批風險重估,行業整體風險上行
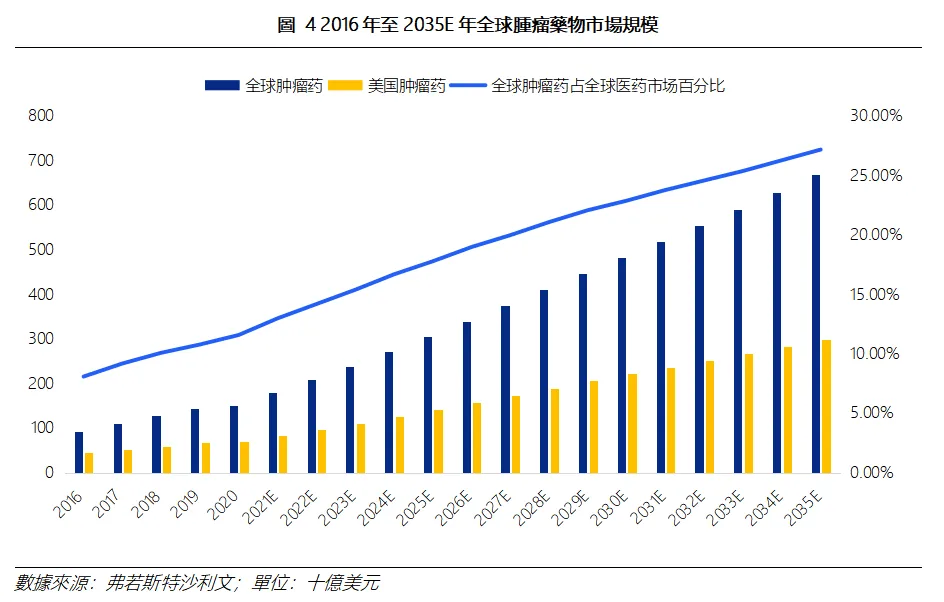
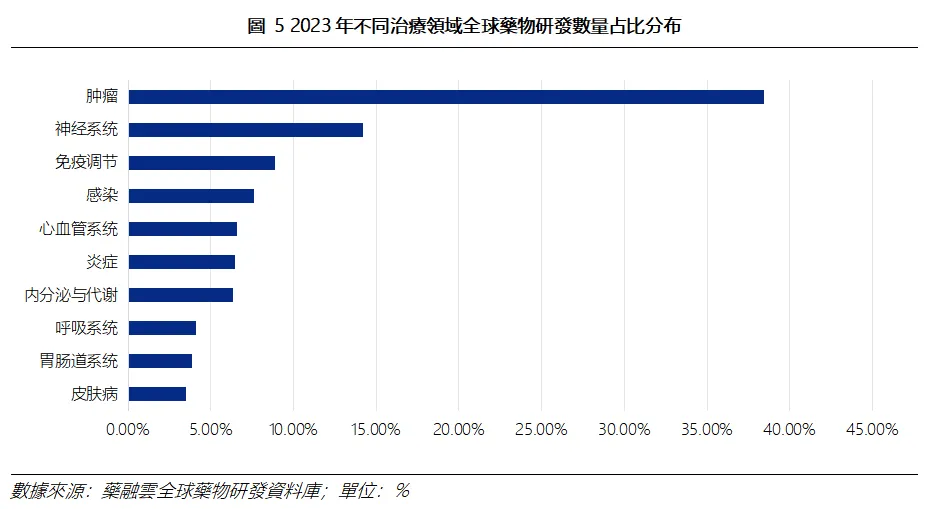
弗若斯特沙利文預測,2025年全球腫瘤藥物市場規模為3048億美元,2035年預計增長至6704億美元,2025-2035年複合年增速達到8.20%;占全球醫藥市場比例有望自2025年的17.80%抬升至2035年的27.20%。
同時,根據藥融雲全球藥物研發資料庫統計,按治療領域分類,全球腫瘤藥物的研發熱度最高。截至2023年4月12日,全球腫瘤藥物合計9.4萬個,占比38%,比例最高。
美國塔夫茨藥物研發中心(Tufts CSDD)指出,作為創新藥領域投入最高、週期最長、風險最大的賽道,腫瘤藥的單藥研發成本(行業均值)約26億美元(包含機會成本)。Tufts CSSD在其2014年及2016年的系列研究中,以每年約7%—11%的資本成本對整個研發週期的資金佔用進行了資本化時間成本折算。若不含機會成本,新藥研發的直接支出中位數約為6.48億美元(區間1.573億—19.508億美元),其中腫瘤藥的直接研發成本中位數約為6.5億—8.5億美元級別。同時,DiMasi等(2016, Journal of Health Economics)基於2003—2011年間的臨床開發成功率的研究,發現腫瘤藥從I期到獲批(整體臨床到上市)的成功率約為11.8%—12.1%。本次 ODAC 否決進一步放大了全鏈條不確定性,對大型藥企、中小型 Biotech 形成差異化衝擊。
審批底層邏輯:從“數據達標” 轉向 “臨床獲益有意義”。過往FDA 對晚期腫瘤藥物審評,常接受替代終點(如 PFS 無進展生存期)作為核心獲批依據,只要數據具備統計學顯著性,獲批概率較高。阿斯利康事件或徹底扭轉這一邏輯。ODAC 明確提出,Camizestrant 僅延長 PFS,但缺失長期總生存期(OS)這一硬終點數據,無法證明 PFS 改善能轉化為患者長期生存獲益。據Targeted Oncology報導,ODAC否決的兩大核心理由即為“對SERENA-6試驗設計的擔憂與缺乏OS數據”。
FDA在2025年12月已通過政策調整推動加速批准後確證性試驗的強制性,以及對替代終點的要求正在全球範圍內收緊。2026年3月Lancet子刊有分析指出,自1992年以來FDA已批准超過200個抗癌藥的加速上市基於替代終點(如PFS),但真實世界數據顯示過半加速批准藥物最終未在OS或生活品質上帶來獲益。這意味著ODAC對camizestrant的審慎態度具有行業全局性背景,並非針對這一特定藥物——ODAC此次投票實際上延續了FDA自2024—2025年以來逐步提升對替代終點審評門檻的長期趨勢。
由此可見,未來腫瘤藥審批有望遵循新標準:統計學顯著只是基礎,必須同時證明療效具備臨床價值(延長生存期、改善生活品質、減少治療負擔等)。僅靠替代終點“好看” 的藥物,很難再順利通關。
大型藥企管線佈局趨於保守,賽道競爭格局重塑;中小型Biotech生存壓力陡增,研發策略被迫回撤。阿斯利康、禮來、羅氏等巨頭均深度佈局SERD(選擇性雌激素受體降解劑)乳腺癌賽道,Camizestrant 是阿斯利康管線中的核心潛力品種。而camizestrant案例並非孤例,2026年FDA也曾拒絕Replimune和Atara的兩項療法申請,理由分別是“試驗設計不充分”和“療效證據不足”。這顯示了該趨勢已在同期發生,與前述拒絕案例共同構成了FDA審評標準正在系統性抬升的監管圖景。在該監管圖景影響下,頭部企業或將全面複盤在研同類項目,重新評估現有試驗終點、數據完整性、治療策略合理性,暫緩部分激進的創新專案推進,優先完善證據鏈。原本白熱化的 SERD 賽道、實體瘤早期干預賽道,短期會進入 “觀望 + 補數據” 階段,企業從 “搶進度” 轉向 “穩證據”,賽道競爭節奏或被動放緩。而中小型生物技術公司是全球創新藥的源頭,但普遍存在現金流緊張、管線單一、抗風險能力弱的特點,和大型藥企形成鮮明對比。若核心候選藥物在 NDA(新藥上市申請)階段被 FDA 顧問組否決,Biotech 極易出現估值暴跌、融資困難、專案終止,甚至公司經營危機。大量中小 Biotech 或將主動規避前沿、高監管風險的創新方向(如分子標誌物早期干預、全新治療範式),轉向監管規則成熟、獲批概率更高的傳統適應症、成熟療法,行業前沿創新的短期活躍度有所下降。
2.2.3. 精准醫療與液體活檢(ctDNA)、伴隨診斷賽道監管趨嚴
Camizestrant 的用藥方案高度依賴ctDNA 液體活檢檢測 ESR1 突變,屬於典型的 “藥物 + 伴隨診斷” 捆綁模式。藥物審批遇阻,直接連鎖影響上游診斷技術的臨床落地與監管規則。
ctDNA 液體活檢的臨床落地節奏放緩。ctDNA 是當下腫瘤精准醫療的核心技術,其核心價值就是 “早於影像學發現腫瘤進展”,行業長期看好其在動態監測、指導治療中的應用。FDA已批准多款液體活檢產品作為伴隨診斷用於臨床,包括Guardant360 CDx(作為固體腫瘤全面基因組分析CDx)、FoundationOne Liquid CDx等。2024年,FDA還批准了Shield用於結直腸癌篩查——這是首個獲批用於癌症早篩的液體活檢產品。此外,Quest Diagnostics的Haystack MRD檢測於2025年8月獲得FDA突破性設備認定。不過,需要注意的是,雖然ctDNA檢測技術本身已被FDA批准(如Guardant360 CDx),但“依據ctDNA指導治療時機切換的臨床策略”這一範式尚未被認可。本次阿斯利康事件證明,診斷技術的先進性,無法彌補臨床療效證據的缺失。在沒有充足臨床數據支撐的前提下,FDA 不會為 “ctDNA 監測 + 提前干預” 的組合模式放行。短期內,全球範圍內基於 ctDNA 指導腫瘤治療時機調整的臨床研究、產品申報或趨於謹慎,該技術從科研場景全面走向常規臨床應用的進程有被延緩的風險。
伴隨診斷(CDx)進入 “捆綁式從嚴審批” 階段。作為長期實行的正式政策,FDA要求伴隨診斷(CDx)必須在相關藥物上市前獲得FDA的批准、許可或授權,其必須表現出穩健的分析有效性(準確可靠檢測生物標誌物)、臨床有效性(預測患者對治療的反應)和臨床實用性(適當指導患者管理並改善患者結局)。ESR1 突變檢測試劑是 Camizestrant 的必需伴隨診斷,藥物能否獲批,和診斷產品的準確性、臨床實用性深度綁定。
FDA 明確要求藥物療效證據 + 伴隨診斷產品價值必須形成完整閉環證據鏈,二者缺一不可。2025年12月,FDA在《聯邦公報》發佈擬議令,計劃將癌症靶標藥物的伴隨診斷與相關分子檢測從III類(需要上市前批准PMA)降為II類(通過510(k)路徑)。這一重要動向說明,FDA在提高藥物審批標準的同時,也在同步優化伴隨診斷的審批路徑靈活性,兩者共同構成了監管體系的協同進化,而非單向趨嚴。未來藥企佈局 “創新藥 + 專屬伴隨診斷” 組合時,不能只驗證藥物療效,還需同步驗證診斷試劑的檢測精度、適用人群、臨床指導價值,藥物與診斷企業都將面臨雙重審評壓力。
2.2.4. 正向行業變革:倒逼全行業提升科學嚴謹性(長期利好)
短期審批風險上升、創新節奏放緩的背後,是行業長期發展的“糾偏”。本次阿斯利康事件暴露了過去部分研發專案 “重概念創新、輕臨床價值;重短期數據、長期證據缺失” 的問題,也推動行業建立更規範的研發體系。
臨床試驗設計前置優化,從“事後補救” 變為 “事前合規”。過去不少藥企的模式是先完成臨床試驗,拿到數據後再對接監管要求。如今轉變思路,在試驗方案設計之初就對標FDA 最新審評標準,比如:優先規劃 OS(總生存期)等長期硬終點,不再單純依賴 PFS 等替代終點;提前打磨對照組、交叉用藥規則,規避試驗設計缺陷;從源頭論證治療策略的臨床意義,而非申報階段再補充解釋。
藥企與監管機構的早期溝通成為標配。針對前沿療法、全新治療範式,藥企會主動在臨床前、I/II 期臨床試驗階段就與 FDA 召開正式溝通會議,提前確認試驗終點、設計方案、審評尺度,提前排查風險。“先溝通、再研發” 將成為創新藥,尤其是 first-in-class(全球首創)藥物的常規操作,大幅降低後期申報被否決的概率。FDA的Advancing RWE專案本身已包含sponsor在提交RWE提案前進行早期溝通的機制——2025年12月15日FDA發佈的相關指南明確了sponsor可通過該程式提交初始會議請求,這為未來更多新產品提前溝通提供了正式制度化路徑。
真實世界證據(RWE)的應用價值與行業權重提升。本次事件暴露出傳統III 期臨床試驗的天然短板:OS 等長期終點數據讀出週期極長(本次預計還需 2 年),無法快速驗證藥物長期價值。為彌補這一缺陷,料想全球藥企、CRO 機構有望加速佈局真實世界研究,依託真實臨床場景數據,補充長期生存期、不同人群療效、長期安全性等資訊,作為傳統臨床試驗的有力佐證。未來真實世界證據,會成為腫瘤新藥申報中彌補數據短板、增強說服力的重要工具。
阿斯利康Camizestrant 被否決,本質是FDA 對全球腫瘤創新藥行業的一次監管糾偏。FDA一方面不再為 “單純的技術創新、模式創新、概念創新” 背書,患者真實臨床獲益成為第一審批標準;另一方面,對 “無監管先例” 的全新治療範式、試驗設計,執行最嚴格的證據要求;同時,藥物、伴隨診斷、前沿檢測技術形成一體化監管,全鏈條標準同步抬升。
短期來看,本次事件或迫使腫瘤創新藥審批風險上行,前沿賽道(SERD、ctDNA 伴隨診斷、腫瘤早期干預)節奏放緩,中小 Biotech 承壓。長期來看,FDA淘汰 “偽創新” 專案,倒逼行業回歸臨床本質,推動臨床試驗、監管溝通、證據體系走向規範化、科學化,利於全球創新藥行業健康可持續發展。
投資評級說明:
買入:預期未來6-12個月內上漲幅度在15%以上;增持:預期未來6-12個月內上漲幅度在5%-15%;中性:預期未來6-12個月內變動幅度在-5%-5%;E-mail:ryan.chan@eddid.com.hkE-mail:gang.chen@eddid.com.hkE-mail:liuzongwu@eddid.com.hkE-mail:sunny.hou@eddid.com.hkE-mail:brook.zhang@eddid.com.hk負責撰寫本報告的全部或部分內容之分析員,就本報告所提及的證券及其發行人做出以下聲明:(1)發表於本報告的觀點準確地反映有關於他們個人對所提及的證券及其發行人的觀點;(2)他們的薪酬在過往、現在和將來與發表在報告上的觀點並無直接或間接關係。此外,分析員確認,無論是他們本人還是他們的關聯人士(按香港證券及期貨事務監察委員會操作守則的相關定義)(1)並沒有在發表研究報告30日前處置或買賣證券;(2)不會在發表報告3個工作日內處置或買賣本報告中提及的證券;(3)沒有在有關香港上市公司內任職高級人員;(4)研究團隊成員並沒有持有有關證券的任何權益。本報告內所提及的任何投資都可能涉及相當大的風險。報告所載數據可能不適合所有投資者。艾德證券期货不提供任何針對個人的投資建議。本報告沒有把任何人的投資目標、財務狀況和特殊需求考慮進去。而過去的表現亦不代表未來的表現,實際情況可能和報告中所載的大不相同。本報告中所提及的投資價值或回報存在不確定性及難以保證,並可能會受目標資產表現以及其他市場因素影響。艾德證券研究部建議投資者應該獨立評估投資和策略,並鼓勵投資者諮詢專業財務顧問以便作出投資決定。本報告包含的任何資訊由艾德證券研究部編寫,僅為本公司及其關聯機構的特定客戶和其他專業人士提供的參考數據。報告中的資訊或所表達的意見皆不可作為或被視為證券出售要約或證券買賣的邀請,亦不構成任何投資、法律、會計或稅務方面的最終操作建議,本公司及其雇員不就報告中的內容對最終操作建議作出任何擔保。我們不對因依賴本報告所載資料採取任何行動而引致之任何直接或間接的錯誤、疏忽、違約、不謹慎或各類損失或損害承擔任何的法律責任。任何使用本報告息所作的投資決定完全由投資者自己承擔風險。本報告基於我們認為可靠且已經公開的資訊,我們力求但不擔保這些資訊的準確性、有效性和完整性。本報告中的資料、意見、預測均反映報告初次公開發佈時的判斷,可能會隨時調整,且不承諾作出任何相關變更的通知。本公司可發佈其他與本報告所載資料及/或結論不一致的報告。這些報告均反映報告編寫時不同的假設、觀點及分析方法。客戶應該小心注意本報告中所提及的前瞻性預測和實際情況可能有顯著區別,唯我們已合理、謹慎地確保預測所用的假設基礎是公平、合理。艾德證券研究部可能採取與報告中建議及/或觀點不一致的立場或投資決定。本公司或其附屬關聯機構可能持有報告中提到的公司所發行的證券頭寸並不時自行及/或代表其客戶進行交易或持有該等證券的權益,還可能與這些公司具有其他相關業務聯繫。因此,投資者應注意本報告可能存在的客觀性及利益衝突的情況,本公司將不會承擔任何責任。本報告版權僅為本公司所有,任何機構或個人於未經本公司書面授權的情況下,不得以任何形式翻版、複製、轉售、轉發及或向特定讀者以外的人士傳閱,否則有可能觸犯相關證券法規。本報告乃由艾德證券期貨有限公司(「艾德證券期貨」)於香港提供。艾德證券期貨是香港證券及期貨事務監察委員會(「香港證監會」)持牌法團,及受其監管之香港金融機構。報告之提供者,均為香港證監會持牌人士。投資者如對艾德證券期貨所發的報告有任何問題,請直接聯絡艾德證券期貨。本報告作者所持香港證監會牌照的牌照編號已披露在作者姓名旁。對部分司法管轄區或國家而言,分發、發行或使用本報告或會抵觸當地法律、法則、規定,當中或包括但不限於監管相關之規例、守則及指引。本報告並非旨在向該等司法管轄區或國家的任何人或實體分發或由其使用。







 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
